


Friday SNPpets
-
This week we’ve got DNA in the gig economy and for sports fans (?), new
software resources for virus and lipids, a handy collection of cancer
genomics pape...
Há 6 anos
Tightly targeted cancer therapy receives marketing approval
Eventually, Clay Siegall got used to doors slamming in his face. When the cancer researcher decided to create a company that would fight cancer using weaponized antibodies, investors were sceptical. "We contacted 35, then 40, then 45 venture-capital companies," he says. "We got turned down over and over and over."
Siegall and his partners kept trying, and 13 years later the investors who eventually bet on Siegall's company, Seattle Genetics, of Bothell, Washington, are getting their reward. On 19 August, the US Food and Drug Administration (FDA) approved the company's lead therapy, an antibody engineered to deliver a poisonous payload directly into lymphoma cells. The hope is that such antibodies, called antibody–drug conjugates, will sidestep the punishing toxicities of classical chemotherapies, which run loose in the bloodstream and kill healthy cells in addition to their targets.
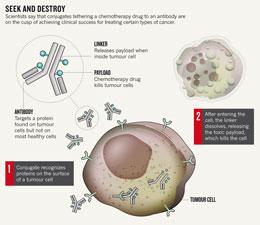 Click for full graphic
Click for full graphicTim Illidge, an oncologist at the University of Manchester, UK, says that the approval is a "game-changer" for a promising class of drugs that has struggled to gain a foothold since it was first described in a 1964 Nature paper1 (see 'A long time coming'). "We're essentially in a renaissance of the antibody–drug conjugate," he says.
Unembellished, or 'naked', antibodies are already used to treat cancer because of their unparallelled ability to target proteins found on the surface of tumour cells. Their high profit margins and strong patent protections have pharmaceutical companies clamouring for more. Siegall says that Seattle Genetics toyed with naked antibodies, too. "But by and large," he says, "most naked antibodies just don't have a strong, potent ability to knock out tumour cells."
Enter antibody–drug conjugates, which do have that knock-out punch. Their power comes from their payloads: lethal drugs tethered to the antibody that remain harmless until the conjugate releases them inside cancer cells (see 'Seek and destroy').
But for decades, developers have struggled to get the crucial elements to work together: the antibody, the drug it carries and the linker that binds the two. Seattle Genetics' conjugate, Adcetris (brentuximab vedotin), seems to have overcome the hurdles; it combines a synthetic poison called vedotin with an antibody that targets CD30, a protein found on many lymphoma cells.
In July, an FDA advisory committee voted unanimously in support of accelerated approval for Adcetris after Seattle Genetics reported that 94% of the 102 people with Hodgkin's lymphoma in a trial of the treatment saw their tumours shrink, and 73% achieved partial or complete remission. "This drug is wildly active," said panellist Mikkael Sekeres, an oncologist at the Cleveland Clinic in Ohio, after the vote.
The accelerated approval means that the drug can now be prescribed by doctors while Seattle Genetics conducts follow-up clinical studies. Mark Monane, a senior analyst at the investment-banking firm Needham & Company in New York, predicts that the drug will bring in up to US$400 million a year in sales.
“It’s like a game of whack-a-mole. You knock out one toxicity and another shows up.”
Janice Reichert, an analyst at the Tufts Center for the Study of Drug Development in Boston, Massachusetts, expects more approvals to follow. Between 2000 and 2005, only six antibody–drug conjugates entered the clinic for the first time, she says. From 2005 to 2009, 15 more joined their ranks. Now, 25 are currently in cancer clinical trials — more than at any other time. Two have reached late-stage clinical trials: trastuzumab emtansine, a breast-cancer therapy jointly developed by biotechnology firms Genentech, based in South San Francisco, and ImmunoGen in Waltham, Massachusetts; and inotuzumab ozogamicin, a lymphoma therapy developed by Pfizer in New York.
Pitfalls remain. The only other conjugate to ever win accelerated approval from the FDA, a different version of Pfizer's lymphoma therapy, was pulled from the market last year after further tests showed that the drug offered no benefits over standard chemotherapy. Many blame the failure on a linker that fell apart in the bloodstream, boosting toxicity and limiting the dose that could be used. Seattle Genetics had to pull one of its antibodies from clinical trials for similar reasons, says Siegall. As a result, the company developed a linker that is degraded by the enzymes that are most active inside the cell.
ImmunoGen, Seattle Genetics' main competitor, has struggled to find the right drug to couple to its antibodies. For years, the company attempted to use ricin, a toxin produced by castor beans. But ricin triggered a dangerous immune response. ImmunoGen now steers clear of complex proteins and picks small-molecule poisons that are less likely to attract the attention of the immune system.
A lingering problem for the field is a lack of control over how many drug molecules attach to each antibody. More control would help standardize each dose and lessen the potential toxicity of the treatment. A method developed by researchers at Genentech, a subsidiary of Swiss drug firm Roche, seemed to have conquered the problem but has not yet been tested clinically2. Unexpected toxicities led the company to shelve the technique for the time being, says Paul Polakis, Genentech's director of cancer targets. "It's like a game of whack-a-mole," he sighs. "You knock out one toxicity and another shows up."
ADVERTISEMENT

Although antibody engineers still have work to do in optimizing the design, the approval of Adcetris means they have a bellwether to watch. The improving fortunes in the field, which culminated in last week's approval, have finally brought Siegall the investor attention that eluded him for so long. Seattle Genetics has partnered with 11 outside firms, bringing in $150 million in new capital. More than a quarter of that was raised in the past year. "I'm happy to say I no longer have to convince investors that this is a productive field," he says.
All of this leaves those who have followed the technology marvelling at its reversal of fortune. "When I first became interested in the field in the early 1990s, there was a lot of despondency," says Illidge. "And now look at it. Everything has changed."
A growing number of studies point to rapamycin as a pharmacological compound that is able to provide neuroprotection in several experimental models of neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, Huntington's disease and spinocerebellar ataxia type 3. In addition, rapamycin exerts strong anti-ageing effects in several species, including mammals. By inhibiting the activity of mammalian target of rapamycin (mTOR), rapamycin influences a variety of essential cellular processes, such as cell growth and proliferation, protein synthesis and autophagy. Here, we review the molecular mechanisms underlying the neuroprotective effects of rapamycin and discuss the therapeutic potential of this compound for neurodegenerative diseases.
http://www.nature.com/nrn/journal/v12/n8/full/nrn3068.html
Acontecerá nos dias 12-15 de outubro na cidade de Floránopolis - SC o 7th International Conference of the Brazilian Association for Bioinformatics and Computational Biology (AB3C) e o 3rd International Conference of the IberoAmerican Society for Bioinformatics (SoIBio). X-meetig 2011 (AB3C / SolBio).
O encontro contará com os palestrantes brasileiros Ana Tereza Vasconcelos (LNCC) e Aristóteles Góes-Neto (UEFS), bem como os palestrantes internacionais Ana Teresa Freitas (INESC Lisboa), Robin Haw (Ontario Inst. Cancer. Res.), Erik Lindahl (Stockholm University), Arthur J. Olson (Scripps Res. Inst.), entre outros.
As inscrições serão no período de 12 de setembro a 15 de outubro de 2011 no site http://www.x-meeting.com/ . A submissão de artigos será no período de 21 de junho a 1 de setembro de 2001 no site. Para maiores informações visite o site do x-meeting.
Nature Reviews Drug Discovery 10, 243-245 (April 2011) | doi:10.1038/nrd3413
The US approval of Human Genome Sciences (HGS) and GlaxoSmithKline's belimumab (Benlysta) on 9 March ended a 50-year drought in the introduction of new lupus drugs. The two firms have blazed a path from mRNA to medicine in 15 years, and analysts are now predicting peak US sales of over US$2 billion annually for the monoclonal antibody (mAb).
Nature Reviews Drug Discovery 10, 307-317 (April 2011) | doi:10.1038/nrd3410
Juswinder Singh, Russell C. Petter, Thomas A. Baillie & Adrian Whitty
Covalent drugs haveproved to be successful therapies for various indications, but largely owing to safety concerns, they are rarely considered when initiating a target-directed drug discovery project. There is a need to reassess this important class of drugs, and to reconcile the discordance between the historic success of covalent drugs and the reluctance of most drug discovery teams to include them in their armamentarium. This Review surveys the prevalence and pharmacological advantages of covalent drugs, discusses how potential risks and challenges may be addressed through innovative design, and presents the broad opportunities provided by targeted covalent inhibitors.
Um biofármaco que acaba de ser desenvolvido por pesquisadores da Universidade Federal do Rio de Janeiro (UFRJ) - e que já teve seu pedido de patente depositado no Instituto Nacional da Propriedade Industrial (INPI) - pode representar um novo caminho para tornar o tratamento do diabetes mais eficaz. O medicamento, produzido no Laboratório de Biotecnologia Farmacêutica da universidade (BiotecFar), é baseado em um sistema de liberação prolongada de amilina humana e tem como objetivo oferecer aos diabéticos um melhor controle da glicemia.
Trocando em miúdos, a amilina é um hormônio produzido naturalmente no pâncreas (pelas células beta), que desempenha um papel fundamental em diversos órgãos, inclusive para equilibrar os níveis de glicose. Este hormônio é cosecretado com a insulina, exercendo conjuntamente papéis importantes na regulação metabólica. "Mesmo pacientes diabéticos que fazem uso da insulina possuem dificuldades de controle dos níveis de glicose no sangue", explica o professor da UFRJ Luís Maurício Lima, coordenador do projeto que teve início em 2009.
Atualmente, o tratamento para diabetes leva em conta apenas a reposição de insulina, deixando de lado a reposição da amilina. Isso ocorre devido à dificuldade de desenvolver medicamentos a base de amilina humana, que é bastante insolúvel. "Ao contrário da insulina, que é livremente solúvel, a amilina humana tem um problema de agregação protéica, que inclusive é causa de diabetes amiloidogênica e ainda a razão da dificuldade de usar o hormônio natural terapeuticamente", afirma o farmacêutico.
Para contornar esse obstáculo e desenvolver o novo medicamento, os pesquisadores do BiotecFar/UFRJ recorreram a um minucioso trabalho de nanobiotecnologia farmacêutica. No laboratório, eles encapsularam nanopartículas de amilina humana em partículas poliméricas biocompatíveis. Por serem tão pequenas, elas podem ser facilmente administradas por injeção subcutânea ou intramuscular e por terem como base polímeros biocompatíveis são naturalmente degradadas e eliminadas pelo organismos. Apesar de continuarem insolúveis, formam um depósito que vai se degradando aos poucos no local de aplicação. "Produzimos nanopartículas contendo amilina humana de 200 nanômetros, que é uma medida equivalente a cerca de um milionésimo de metro", conta Lima.
Uma vantagem do uso de nanopartículas é a liberação contínua e lenta da amilina humana. Esta característica permite que as aplicações de amilina humana, sejam por injeções intramuscular ou subcutânea, não precisem ser diárias. "Esse detalhe é importante para manter a qualidade de vida do paciente diabético que já recebe doses diárias de insulina. Assim, o paciente não precisaria receber mais injeções diárias, de análogos solúveis de amilina, o que tornaria o tratamento mais desconfortável e menos próximo ao fisiológico por não se tratar da amilina humana. Podemos programar aplicações semanais ou até mensais", destaca o professor. "A ideia é que a reposição de amilina humana seja um tratamento complementar à reposição de insulina, para potencializar o controle da glicemia", completa.
Leia a matéria completa no site: www.faperj.br.
(Agência Faperj)
If dogma dictates that proteins need a structure to function, then why do so many of them live in a state of disorder?
Keith Dunker's life is a mess. His desk is so swamped with books, old chocolate bars, half-reviewed manuscripts, pens, coke bottles and — somewhere — a stray sock, that he ends up printing papers again rather than wading in to find the original. "I'm so disorganized," he crows, "some people have called me Dr Disorder." But he remembers with great precision the moment that disorder invaded his scientific life. It was 15 November 1995, at 12:40 p.m., halfway through a seminar by crystallographer Chuck Kissinger, at Washington State University in Pullman, where Dunker was then a biochemist. Dunker was staring at a slide showing the atomic structure of calcineurin, an enzyme targeted by immunosuppressive drugs. What caught his attention wasn't the intricate structure but something missing from it: a dotted line representing a string of amino acids with a position too variable to be determined by X-ray crystallography, as the rest of the protein had been. And Kissinger was insisting that this loop had to remain flexible for calcineurin to serve its crucial function in the human immune system.
"It hit me like a brick," says Dunker: this wayward piece of protein flouted a century of dogma. A central tenet in molecular biology is that the function of a protein depends critically on its fixed three-dimensional structure; by extension, enzymes bind to specific substrates because their shapes match perfectly, as immortalized in the 'lock-and-key' model proposed by chemist Emil Fischer as early as 1894. But this part of calcineurin seemed to disobey these rules, by providing function without structure. Now Dunker was wondering how many other proteins were ignoring the rules too.
To find out, he and his colleagues wrote a bioinformatics program that predicted which protein segments are 'intrinsically disordered' — meaning that they do not fold spontaneously into a unique three-dimensional shape. Today, this and other similar programs predict that about 40% of all human proteins contain at least one intrinsically disordered segment of 30 amino acids or more, and that some 25% are likely to be disordered from beginning to end1. This part of the protein universe had largely been ignored because disordered protein segments impede crystal formation — a prerequisite for X-ray diffraction, the predominant way structures are deduced — and structural biologists clip them out whenever they can.
Today, though, "the recognition of disorder has grown dramatically", Peter Wright, a protein biophysicist at the Scripps Research Institute in La Jolla, California, told the American Association for the Advancement of Science meeting in Washington DC last month. A large part of that recognition has come from studies using nuclear magnetic resonance (NMR) spectroscopy, which allows researchers to determine the structures of small proteins even as they twist and turn in solution. Such work has shown that disorder can actually be essential to function by helping a signalling protein to recognize and react to a protein partner, or by allowing a regulatory protein to interact with multiple targets. Still, says Wright, "that hasn't got through to the textbooks".
Many structural biologists see no need for revision. "My mantra has been: function requires structure," declares Tom Steitz, a crystallographer at Yale University in New Haven, Connecticut. "Some flexibility can be required, it may be an essential part of the assembly process, but it's not interesting until the proteins get to do their job." Critics argue that the computer programs predicting high levels of disorder are fundamentally flawed because they identify proteins that are well-known to become perfectly ordered — and to crystallize — when bound to their proper molecular partners. They say that unfolded protein chains cannot persist for long in living cells, and some want the concept of intrinsically disordered proteins to be ditched altogether. That seems unlikely. Data are fast accumulating from all fronts — biophysics, bioinformatics and cell biology — in support of widespread disorder, and disorder aficionados are calling for a complete reassessment of the structure–function paradigm. "Biology uses disorder to bring about its various functions," Wright says.
Since the late 1950s, newly made proteins have been assumed to fold up immediately and spontaneously into a unique three-dimensional shape — their most energetically stable conformation and the only functional one2. The few proteins known to remain unfolded "were pointed to as oddities", Wright says. But that started to change in 1999, when Wright and fellow NMR spectroscopist Jane Dyson, also at Scripps, wrote a review3 pointing to the growing collection of proteins that seemed to function despite their disordered state. It has been "the big-dog paper in the field", says Dunker, now at Indiana University School of Medicine in Indianapolis.
One burning question, then and now, is how a protein can function if it has no fixed shape. "We all accept flexibility," says structural biologist Joël Janin at the CNRS Laboratory of Enzymology and Structural Biochemistry at Gif-sur-Yvette, France. "The question is: how can you get recognition with flexibility?" The whole concept of disorder seems incompatible with the lock-and-key model. You might as well try to open the door with cooked spaghetti.
In 2007, postdoc Kenji Sugase in Wright's lab found an answer: the spaghetti uses the lock to mould itself into the shape of the key, rather than forming the key beforehand. Sugase focused on CREB, a gene-regulatory protein involved in many processes including learning and memory. Once bound to DNA, CREB also needs to recognize and bind a protein partner called CBP before it can switch on the gene. But the part of CREB involved with CBP enters the game in a disordered state. How could a thing like this possibly work?
To find out, Sugase developed the equivalent of a super-fast NMR camera so that he could capture frequent snapshots of CREB's wriggling chain, at atomic resolution, as it tested out points of contact within itself and with CBP. What he saw was that several bonds had to form cooperatively within CREB and with CBP for the whole complex to snap into shape4. That's exactly how the average 'globular' protein folds: its internal segments need to establish long-range chemical bonds with one another, to pull the whole thing suddenly into shape2. CREB forms such interactions externally, by bonding to CBP — and if this bonding is just a little weaker, then the key cannot form and there is no binding with the lock at all. Wright and his colleagues think that disorder is therefore advantageous because it allows CREB to partner with CBP more exclusively than a rigid protein would. And Wright thinks that this type of process allows many signalling proteins to engage in speedy yet selective interactions.
The structure–function mantra took an even bigger hit recently from a protein with parts that never seem to fold at all. The Sic1 signalling protein is a key regulator of the cell cycle that puts the brakes on DNA replication until the cell is ready to divide. In 2001, a team led by Mike Tyers, a yeast cell-cycle expert at the University of Toronto, Canada, began unpicking the mechanism of the switch. The group found that when phosphate groups are added to six sites on Sic1 it can then hook up with a second protein, Cdc4, which pushes Sic1 into the cell's protein-disposal pathways5. Once Sic1 is degraded, DNA replication can forge ahead. But unless that degradation occurs at precisely the right time, DNA replication goes haywire and the cell may eventually die. The cell achieves that precision by ensuring that it takes exactly six phosphates to flip the switch, not four or five. But there's a rub: Cdc4 has only one high-affinity binding pocket for a phosphate group. How can Cdc4 'count' up to six with effectively only one finger to count on?
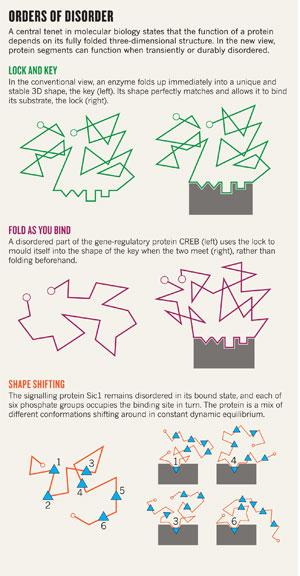 Click for larger version.
Click for larger version.After all attempts to crystallize the Sic1 complex failed, Tyers's team called NMR spectroscopist Julie Forman-Kay, also at the University of Toronto. In 2008, Tanja Mittag, a postdoc in Forman-Kay's lab, showed that Sic1 was disordered6 — not only in its free state but, astoundingly, also when bound to Cdc4. The complex seemed to be a mixture of different conformations shifting around in constant, dynamic equilibrium. And the most stunning part was that each of the six phosphate groups on Sic1 could be found to occupy the single Cdc4 pocket, one after the other, as in a constant dance around the fire (see 'Orders of disorder').
The researchers then developed a computer model and fed it with every scrap of experimental data about the proteins' structures that they could gather7. They concluded that, even though Sic1 is disordered when it is bound, it maintains a rather compact structure, which keeps all the phosphate groups sufficiently close together to form an average electrostatic field that glues Sic1 to Cdc4. Only when six phosphates are present is the glue strong enough for Cdc4 to hold Sic1 close and force-feed it into the cell's disposal machinery. And one reason that Sic1 has to be disordered during all this, the team proposes, is to enable the rather rigid disposal machinery to reach all parts of Sic1 and carpet it with the chemical tags that mark the protein for destruction. It takes a nimble protein indeed to make all these connections at once.
"The result is interesting," says structural biologist Stephen Harrison of Harvard Medical School in Boston, "because interaction motifs are often found more or less repeated along unstructured segments, and the work shows how such multiplicity can function." And Forman-Kay thinks that adopting 'multi-structural' states could allow other proteins to constantly probe and sense signals from many partners at once. This is particularly important for 'hub' proteins, which are central to vast networks of rapidly changing molecular interactions. "There is a complexity people haven't talked about," she says. "These hub proteins need to very rapidly sample the complex cellular environment."
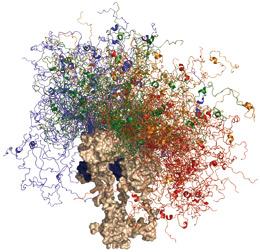 Tumour-supressor protein p53 contains an ordered, globular domain (brown); its disordered segments (colours) help it interact with hundreds of partners.M. BLACKLEDGE/IBS GRENOBLE
Tumour-supressor protein p53 contains an ordered, globular domain (brown); its disordered segments (colours) help it interact with hundreds of partners.M. BLACKLEDGE/IBS GRENOBLEOne extreme example can be seen in the tumour suppressor p53, an extraordinarily well connected hub in multiple signalling networks, and the protein most frequently implicated in human cancer. Part of the explanation for p53's promiscuity seems to lie in its versatile structure, which features every possible conformation from order to disorder. The core domain is globular and binds to DNA and just a few other proteins; its two flanking wings are mostly disordered and can bind to hundreds of signalling partners; and a segment within one wing shows a 'chameleon' status which can flip between four different ordered states, depending on which partner it binds to8. Alan Fersht, a biophysicist and NMR expert at the University of Cambridge, UK, says that he is "absolutely sure" that long parts of p53 remain largely disordered in the cell: "I don't think there's any doubt about that whatsoever."
Yet many researchers question how widespread disordered proteins can be. That is mainly because biochemists, over more than 100 years of preparing tissue extracts, have struggled to prevent proteins from unfolding and tangling into insoluble clumps or getting digested by enzymes called proteases. "It's hard to believe that disordered proteins could produce anything else than a mess," says Janin.
Researchers who study these processes, however, say that such fears are largely irrelevant. In human cells, for example, nonspecific proteases are locked away in compartments called lysosomes, which should allow disordered proteins to survive everywhere else, explains Ulrich Hartl, an expert on protein quality control at the Max Planck Institute in Martinsried, Germany. Disordered proteins should also be protected from aggregation because, unlike globular proteins, they contain few hydrophobic amino acids, which tend to stick together — and are instead rich in 'polar' amino acids that are happy swimming in water.
Hartl thinks that natural selection against aggregation probably gave disordered proteins this particular amino-acid composition — in other words, it is not a signature for disorder per se. And this explains an apparent inconsistency of disorder predictors: that they do not pick internal segments from globular proteins, even though these too are incapable of folding on their own when sliced out of a protein2. The reason is that they are rich in hydrophobic amino acids and so do not show the signature sequence that the predictors detect. It also explains why the predictors select some proteins that are in fact ordered, another point of controversy surrounding these programs: because proteins can lack hydrophobic amino acids for reasons other than disorder. "This makes perfectly good sense," agrees Dunker.
Overall, the numerous programs claim an 80% success rate at predicting whether any individual amino acid in a protein will be surrounded by order or disorder1, as compared to the 50% success rate expected by chance — and crystallographers rely on them to sideline proteins expected to resist crystallization. "Disorder predictors are a massive oversimplification, but they are very useful," says Adam Godzik, at the Burnham Institute in La Jolla, California, and head of the bioinformatics group for the Protein Structure Initiative, a large collaboration that aims to solve large numbers of protein structures.
Nevertheless, debate about the prevalence and importance of disorder has probably slowed progress in the field. DisProt, a database of proteins whose disorder has been established experimentally, contains just over 500 proteins, a number dwarfed by the more than 60,000 structures in the Protein Data Bank1, the database for 3D structures. "The main reason why DisProt is so small," says Dunker, "is because it has taken so many damn years to get it funded." But in the past few years, major consortia aimed at exploring intrinsically disordered proteins have been set up in several countries. Interest in disordered proteins as drug targets is also on the rise because so many of them, like p53, are crucially implicated in disease.
ADVERTISEMENT

Little by little, a fundamentally new picture of the relationships between protein sequence, structure and function is emerging: a continuum running from the most rigid 'lock-and-key' enzymes and molecular machines at one extreme through to durably unstructured spaghetti such as Sic1 at the other, and spanning all degrees of structural ambiguity in between. Figuring out how all these disordered proteins really work is a long way off, if Sic1 is anything to go by: determining its mode of action involved several, often arcane, biophysical techniques, new computer tools and statistical physics theory — plus at least ten years of work by six labs. Multi-structural biology isn't going to be simple.
Still, Dunker, Wright and other doctors of disorder are optimistic. As is Martin Blackledge, an NMR spectroscopist at the Institute of Structural Biology in Grenoble, France, who compares the excitement now to that surrounding the first crystal protein structures in the 1950s. "Every new case is fascinating at the moment," he says. Blackledge looks forward to a day when it might be possible to predict where on the structural continuum a protein segment falls, from its amino-acid sequence — to crack the full code of disorder. "This is exactly what I'm aiming for," he says, "this is my dream."
Perhaps the rules of disorder are needed, before disorder can rule.
Tanguy Chouard is an editor for Nature in London.

